
Polymerizable Lyotropic LC Systems for Templated Nanocomposite Synthesis
Nature is able to construct composite materials with impressive properties by organizing dissimilar components such as mineral and protein into sophisticated nanocomposites. In order to achieve architectural control on this size regime with man-made materials, we pioneered a novel approach that employs cross-linkable lyotropic LCs. We have designed several amphiphilic monomers such as 1 and 2, which self-assemble into an inverted hexagonal (HII) lyotropic LC phase and are able to trap reactive hydrophilic solutions in the ordered nanochannels. Photo-initiated cross-linking locks in the microstructure, and subsequent photolysis or thermolysis converts the reagents trapped within the hydrophilic channels into solid-state "filler" materials (Figure 2).
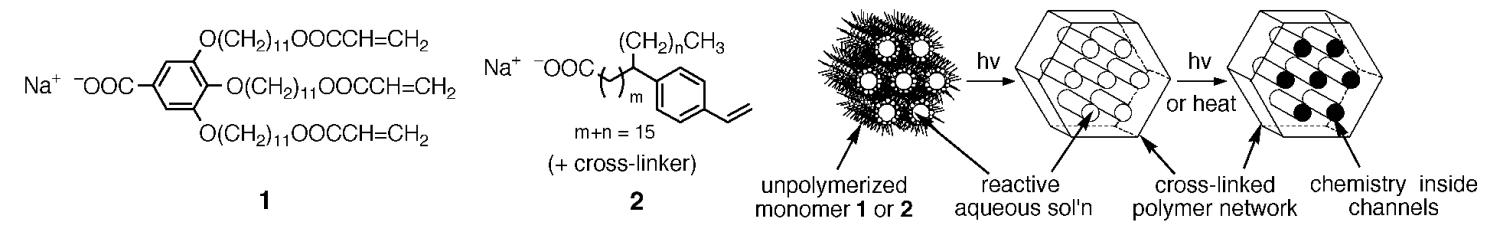
Figure 2. Synthesis of ordered polymer-based nanocomposites using monomer self-assembly.
This synthetic approach yields robust, organic networks possessing hexagonally-packed, monodisperse channels that are approximately 1.2–1.4 nm in diameter with an interchannel spacing of ca. 4 nm, according to X-ray diffraction and TEM imaging studies. We have successfully grown silica and CdS nanoparticles inside the nanostructured matrices. In addition, we have also formed poly(p-phenylenevinylene) (PPV), a conjugated polymer, inside the cross-linked inverted hexagonal phase. (This work was highlighted in the May 19, 1997 issue of Chemical & Engineering News.) The materials formed inside the periodic microdomains exhibit different properties than corresponding materials formed in bulk or in solution. The dimensionally constrained environment apparently limits the degree of conversion of these materials, thereby imparting them with different structures, and consequently different properties, than those obtained in bulk. More recently, nanocomposites containing catalytically active Pd nanoparticles have been synthesized. In these systems, enhanced or modified catalytic behavior is manifested as a consequence of nanoparticle formation in the ordered matrix.
We also have found that the unit cell dimensions and channel diameters of these cross-linked inverted hexagonal phases can be altered systematically in the 1–2 nm range by changing the metal counterion on the monomers, controlling the length and degree of branching of the tails (in the case of homologs of 1), or changing the position of the polymerizable group (as in the case of regio-pure analogs of 2). We are currently examining the effect of channel size on the properties of the "filler" materials formed inside, and expanding the range of organic and inorganic chemistry possible in the nanochannels. In addition, we are investigating the polymerization dynamics and kinetics of these ordered systems.