Quantum Monte Carlo
There are two flavors of QMC, (a) variational Monte Carlo (VMC) and (b) projector Monte Carlo (PMC). VMC starts by proposing a functional form for the wavefunction and then optimizes the parameters of the wavefunction to minmize the energy. At the end of the minimization one obtains a state that has the largest overlap with the ground state of the system, subject to the constraints imposed by the chosen functional form. In PMC one simply performs a imaginary time propagation which is akin to numerically cooling the system down to 0 K. At the end of this process one obtains the ground state energy and wavefunctions. PMC is exact in principle, however, while performing imaginary time propagation one has to use stochastic methods and these suffer from a fundamental problem known as the sign problem. The sign problem typically limits the accuracy with which accurate results can be obtained. In our work we work on both VMC and PMC, in the particular the flavor of PMC that we work on is known as auxiliary field quantum Monte Carlo (AFQMC). We briefly describe the recent developments in AFQMC that have originated from the work done in my group.
Auxiliary field quantum Monte Carlo (AFQMC)
as outlined below.
(a) Accuracy of AFQMC:
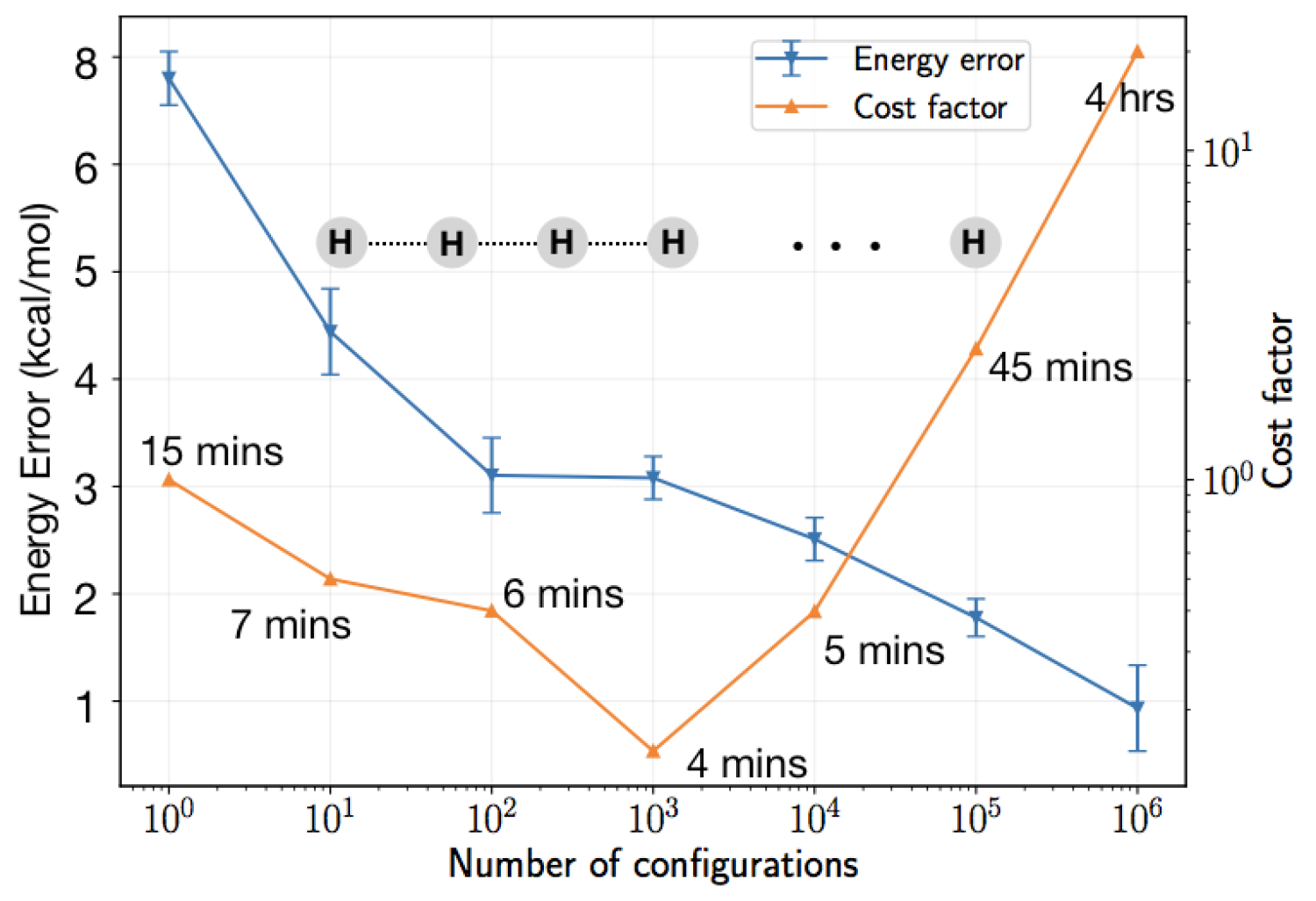
AFQMC can be seen as a method that takes information about the system and a trial state as input and provides the stochastic realization of the ground state and its energy as output. The only uncontrolled approximation made in AFQMC is the phaseless approximation, which relies on the quality of the trial state. In the impractical scenario where the trial state is the exact state, AFQMC yields exact results. However, in practice, a single determinant, such as the Hartree-Fock or DFT wavefunction, is typically used as the trial state. When attempting to enhance the accuracy of the trial state by including a larger number of determinants (Nd determinants), the calculation cost typically increases linearly with the number of determinants (shifting the cost from O(N4) to O(NdN4)), restricting the trial state to only about 100 determinants. To overcome this limitation, my group has developed a novel algorithm that reduces the scaling to O(NdN +N4) instead of O(NdN). This represents a significant improvement in CPU cost and qualitatively alters the types of problems that can be tackled with AFQMC. For instance, as shown in the Figure, for a hydrogen chain increasing the quality of the trial state not only improves the accuracy of AFQMC but remarkably achieves this higher accuracy at a lower computational cost. This is facilitated by the fact that as the quality of the trial state improves, the noise in the simulation decreases. Additionally, because the cost of including a larger number of determinants is relatively inexpensive, higher accuracy can be achieved at a lower cost. Our algorithm has already been taken up by other groups. It was recently used by the Friesner group to investigate a large benchmark comprising more than 300 molecules, including those in the G2, G3, and W4 datasets, predominantly featuring weakly correlated molecules where CCSD(T) exhibits extremely high accuracy. When AFQMC was used with a multiSlater trial employing our algorithm, utilizing fewer than 10,000 determinants (with a computational cost roughly equivalent to AFQMC with a single Slater trial state), the errors were reduced to below those of CCSD(T). This represents one of the first systematic studies involving a large benchmark of weakly correlated molecules where a method has been competitive with CCSD(T), excluding composite methods that themselves rely on CCSD(T) to capture the majority of the correlation energy.
(b) Linear and sub-linear scaling AFQMC:
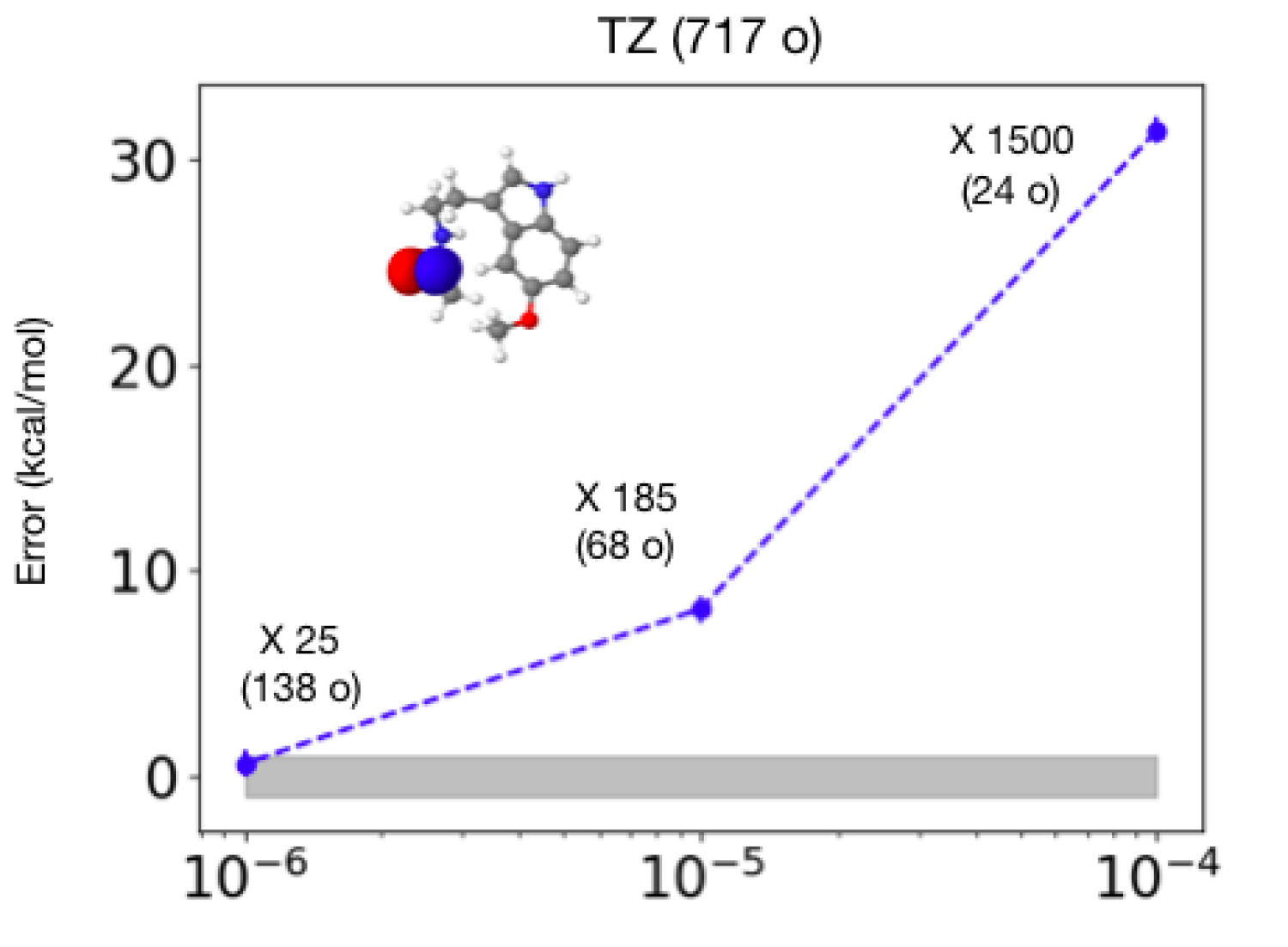
(c) Properties using AFMQC: