By far, the most popular method in quantum chemistry and material science is Density Functional Theory (DFT) because it combines modest cost with relatively high accuracy. The use of DFT blossomed after the introduction of hybrid DFT by Becke in the early 1990s. Although hybrid DFT functionals are routinely used in quantum chemistry, their application in material science, involving periodic systems, is restricted due to the high computational cost. One reason for this is that while Gaussian basis functions provide enormous benefits in molecular systems, this is no longer the case for periodic systems due to the presence of infinite lattice summations.
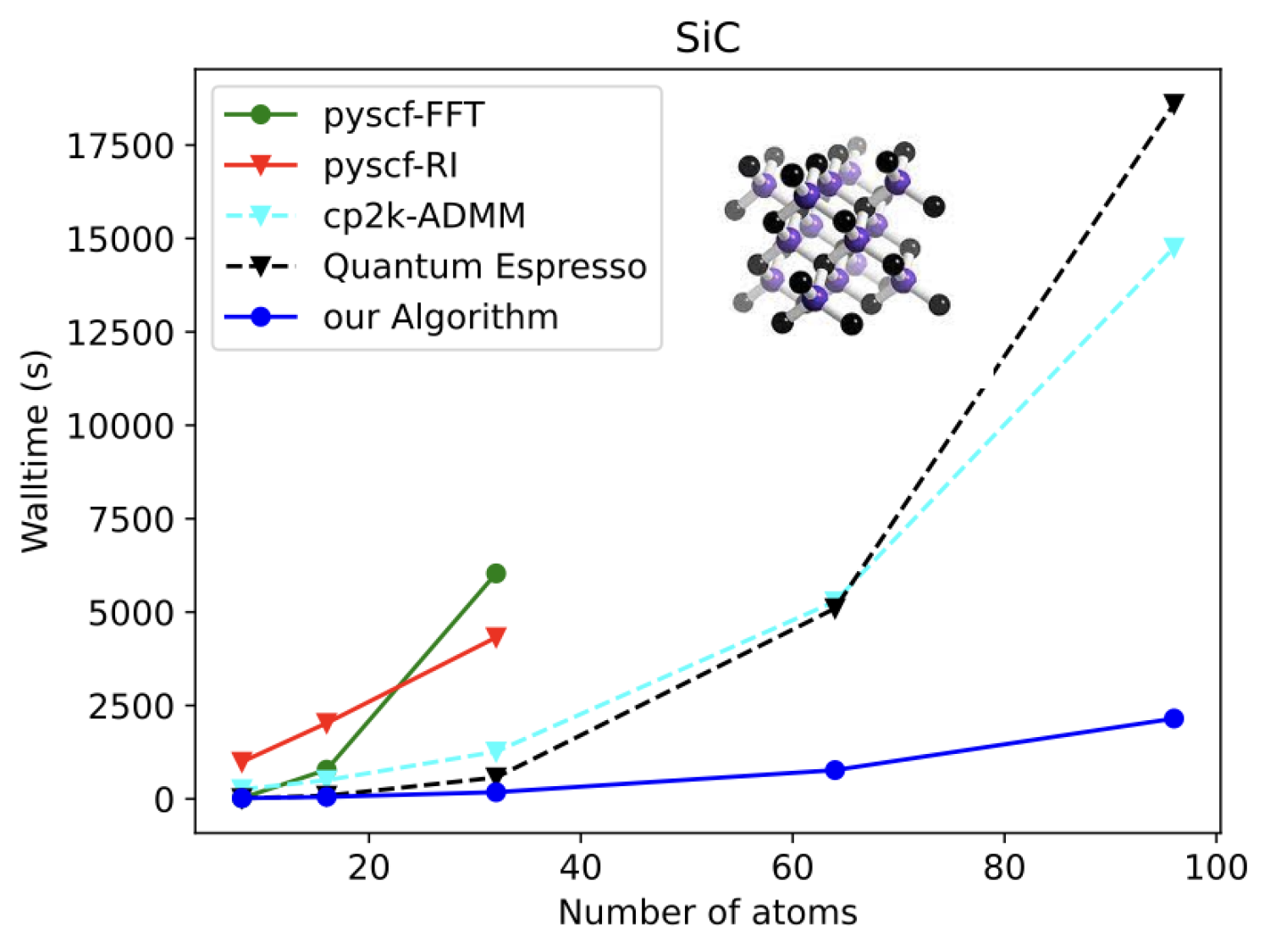
My group started working on hybrid DFT three years ago when we began our collaboration with the Park group, with whom we were trying to understand the properties of conducting metal-organic frameworks (MOFs). For this study, we needed to calculate the band gaps of MOFs for which hybrid DFT would be a suitable theory. However, the cost of hybrid DFT calculations was prohibitively expensive. To overcome this bottleneck, our aim was to develop code that could handle moderately large unit cells (containing about 1000 electrons and 10000 basis functions) on a single compute node. To achieve this, we have paid particular attention to limiting the size of memory usage. Our implementation utilizes several recent developments such as interpolative decomposition (closely related to Martinez’s tensor hypercontraction and similar to Friesner and Neese’s pseudospectral methods), robust fitting that significantly helps reduce errors, occ-RI developed by Head-Gordon (with close ties to Lin Lin’s ACE algorithm). Additionally, we are pioneering the use of multigrid methods to speed up exchange (paper under preparation), which has so far only been used to speed up pure DFT in Cp2K and PySCF. Our recent publication showed a pilot implementation that can outperform several open-source packages, as benchmark data generated by our other collaborators, researcher from Qsimulate software company, demonstrates (Figure above). In our upcoming publication, we have made significant improvements beyond what is presented in the figure and can now perform periodic hybrid DFT calculations for systems with 5000 basis sets on a laptop with only 32 GB of memory, with accuracies competitive with density fitting methods.