
Summary of research contributions

Overview: My research career was devoted to helping to establish the central principals of cellular DNA replication, ranging from mechanisms of nucleotide biosynthesis to the complex interplay of proteins at the replication fork. Approaches applied ranged from chemical biology, biophysics and enzymology to genetics. As with all research, solving one set of problems creates new questions that will be addressed by the next generation of DNA replication enzymologists. Some are briefly stated below, but are discussed in more detail in recent reviews (111, 112). [numbering of references corresponds to publication list.]
Thymidylate synthase mechanism: My graduate work, under Dan Santi’s direction, I showed that thymidylate synthase initiates catalysis by attack of an active site nucleophile on the 5-position of dUMP, creating a carbanion at the 6-position that attacks a carbon at the formaldehyde level of oxidation within 5,10-methylene tetrahydrofolate. These studies showed that a the broadly used cancer chemotherapeutic agent, 5-fluoro dUMP, acts as a suicide inhibitor, trapping the enzyme in a covalent complex (1,2,3,31). Revealing that the reaction was tetrahydrofolate-dependent, led others to develop leucovorin as a chemotherapeutic agent that enhances the effect of 5-fluorouracil (Keyomarsi, Moran (1988) J Biol Chem 263:14402; Grem (2000) Invest New Drugs 18:299).
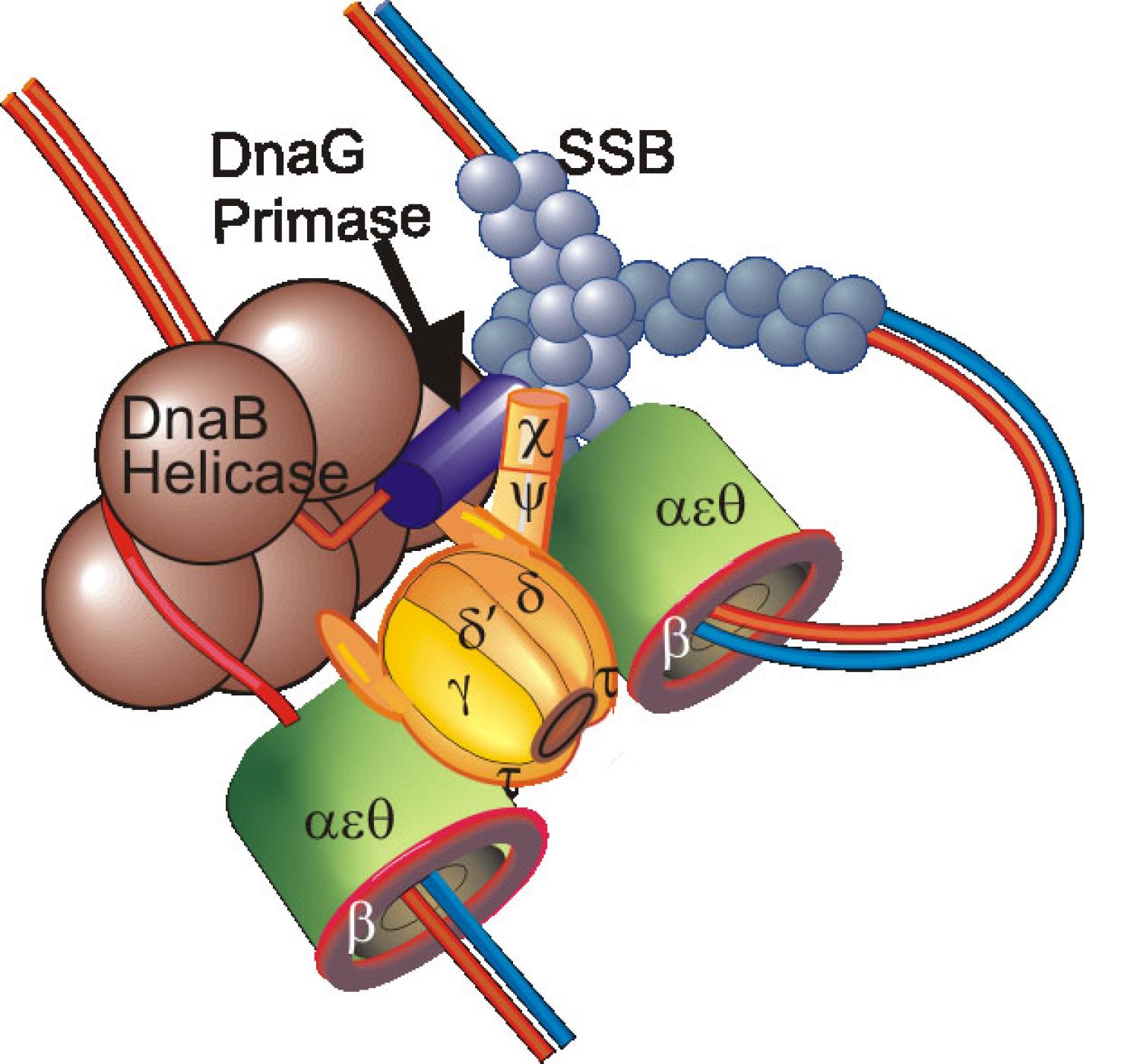
Purification of DNA Polymerase III Holoenzyme and identification of its components: Initially in Arthur Kornberg’s laboratory (4), and then in my own lab (5,8,11,24), I purified the DNA polymerase III holoenzyme (Pol III HE) 10,000-fold and identified most of its subunits. My discovery of tau led to the discovery of a dimeric polymerase, capable of holding the leading and lagging strand polymerase in a complex (11,18). We showed the Pol III HE to be composed of three assemblies: beta2, the DnaX assembly (originally called the gamma-delta complex) and a core polymerase (4,5,24). Bruce Alberts laboratory showed a similar arrangement for the replicase encoded by bacteriophage T4 (Alberts (1987) Philos Trans R Soc Lond 317:395). This tripartite arrangement was later shown, by others, to be conserved in all life forms (O'Donnell, Langston, Stillman (2013) Cold Spring Harb Perspect Biol.).
Identification of a role for beta2 in replication initiation and as a processivity factor: In early work, we developed a purification to yield large quantities of beta2, showed that it was an acidic dimeric protein, that is entered initiation complexes formed by incubating Pol III HE with ATP, and, with Bob Bambara, that beta2 was the key processivity subunit within the Pol III HE (6,7,13,22,23). Collaboratively with Bob Bambara, we first determined the high processivity conferred by auxiliary proteins upon a replicase (7). Using FRET and later photo-crosslinking we showed that beta2 resides two helical turns behind the primer terminus (49,61,62). Later, John Kuriyan’s lab, in collaboration with Mike O’Donnell, determined a structure that established the molecular basis of beta2's processivity (Kong et al. (1992) Cell 69:425). It is a ring like protein that is clamped around DNA in the ATP-dependent step required for forming a processive replication complex (Bowman (2005) FEBS Lett. 579:863).
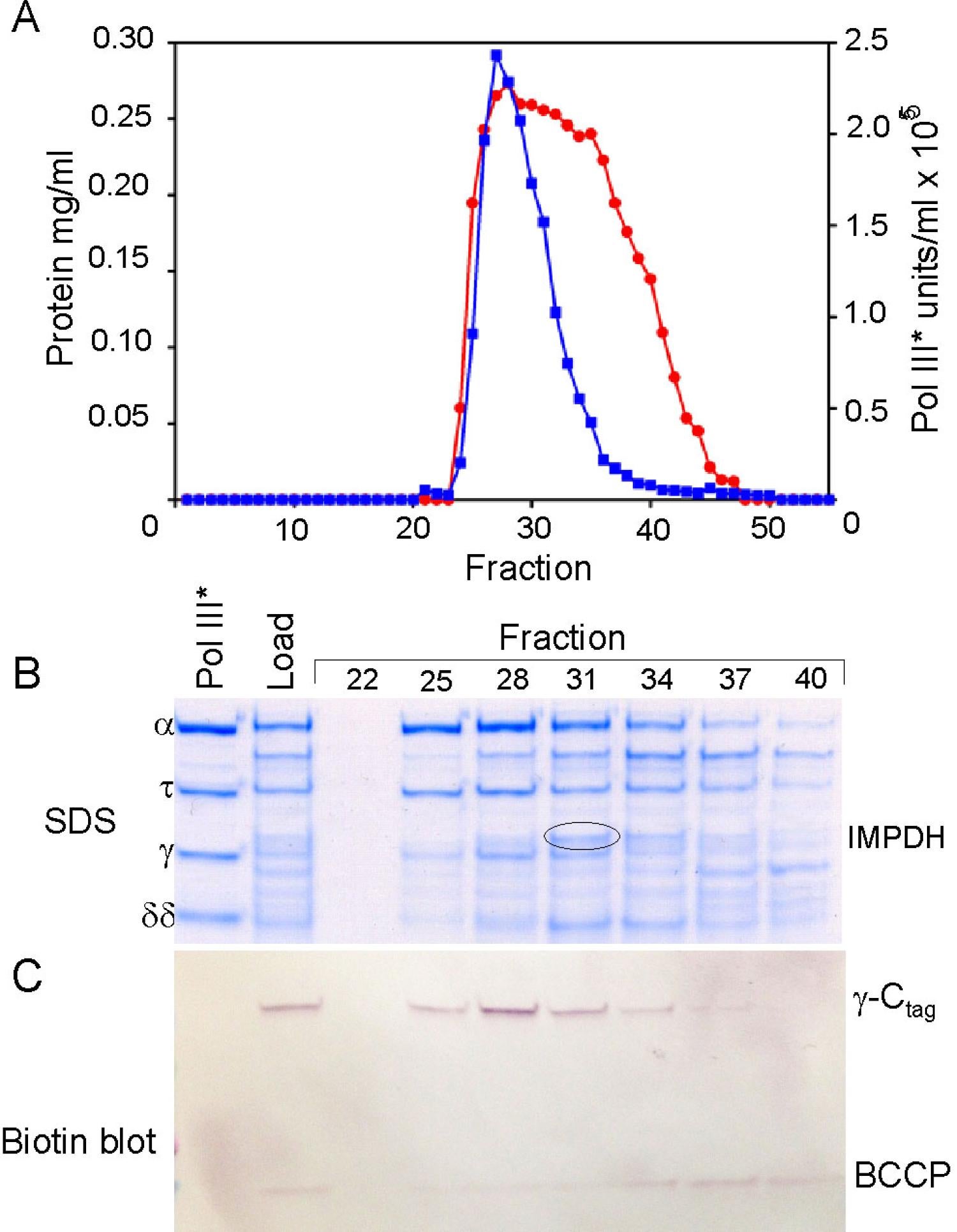
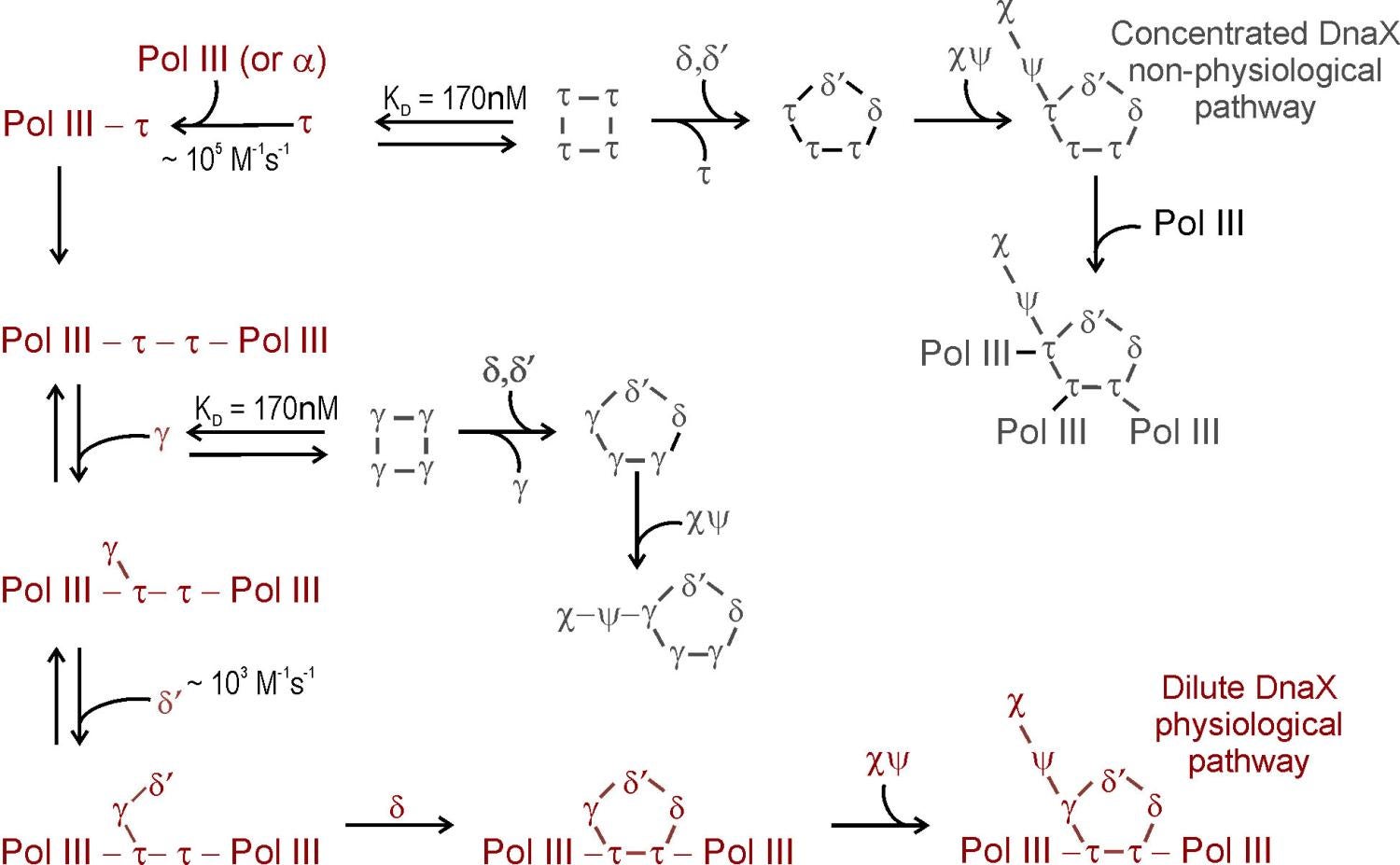
Composition, assembly and function of the DnaX complex: The ATPase of the DnaX complex clamp loader is provided by the tau and gamma subunits, both encoded by dnaX. The shorter gamma product is produced by programmed ribosomal frameshifting (40,41; Blinkowa, Walker (1990) Nucleic Acids Res. 18:1725; Tsuchihashi, Kornberg (1990) Proc Natl Acad Sci 87:2516). Tau contains two extra domains that bind the DnaB helicase and Pol III alpha (86,87). Although we were initially confused by both tau and gamma forming tetramers in solution (65), we established their stoichiometry within the DnaX complex to be three with one subunit each of delta, delta’, chi and psi (65,85). Structural studies later confirmed this stoichiometry (Jeruzalmi (2001) Cell 106:429). Simple mixing of all components of the DnaX complex fails to form complexes containing both gamma and tau (91). Challenges in assembly of a mixed DnaX complex in vitro and the interpretation of whole cell single-molecule experiments led to proposals that Pol III HE contained three taus, thus three associated Pol IIIs (McInerney et al., (2007) Mol Cell 27:527; Reyes-Lamothe et al., (2010) Science 328:498). This led us to formulate an unambiguous test for our earlier conclusions that could have been explained if the gamma found in Pol III HE was generated by proteolysis. We expressed tau from chromosomal dnaX, mutated so that it did not frameshift to produce gamma. Tagged gamma was expressed in trans from an uninduced plasmid so that tagged-gamma levels were physiological. From these cells, purified Pol III HE contained tau and one tagged gamma subunit, unambiguously showing that the two proteins are assembled into the same complex in vivo (130). We have proposed a model where co-assembly requires the low levels of free tau and gamma that would be observed in vivo and the presence of Pol III to steer assembly to Pol III’ that would then sequentially associate with gamma, delta, delta’ and chi psi (111,125).
As shown by cross-linking studies, Pol III HE contains a single copy of gamma in a unique position (83). The asymmetric distribution of tau relative to gamma within Pol III HE may explain the asymmetric behavior of two halves of the enzyme observed in the presence of ATPgammaS and might relate to the issue created by the asymmetric function of the two halves of a dimeric Pol III HE at the replication fork (89). In addition to the established role of tau in binding and stimulating the replicative helicase, tau is required to permit chaperoning of Pol III onto beta2 nascently assembled by the same DnaX complex (108,113). The function of gamma appears to be to avoid the presence of a third copy of Pol III which would outcompete exchange of other polymerases, such as Pol IV, into the replication fork (130)
Coordinated action of the leading and lagging strand replicase and cycling of the lagging strand polymerase, during Okazaki fragment synthesis: An interest throughout my career has been the mechanism that leads to recycling of the lagging strand polymerase during replication and coordination of this event with the leading strand polymerase and replication fork helicase. With Bob Bambara, we showed that the Pol III HE was so processive that a mechanism must exist to cause the release of the lagging strand polymerase during Okazaki fragment synthesis (7). Early studies showed that addition of extra beta2 to reactions greatly accelerated Pol III recycling to another template, suggesting that new beta2 was used with each round of replication (24). We also showed that beta2 was part of the resulting elongation complex, disproving an alternative theory considered at the time that beta2 was only involved in initiation (13). A collaboration with Ken Marians showed that primase was involved in setting the clock for recycling (51). Ken put forward a signaling hypothesis that implicated action of DnaG in triggering recycling of the lagging strand polymerase (51). Also with Marians, we showed that the tau subunit of Pol III HE bound the DnaB helicase at the fork and accelerated its progression (67,3,84,87).
Dissection of the Pol III alpha catalytic subunit revealed an internal domain that was required for beta2 binding (72). A bioinformatics approach later identified a beta binding consensus sequence within the beta2 binding domain, reinforcing our conclusion (Dalrymple et al. (2001) Proc Natl Acad Sci 98:11627). However, our work was challenged and a Pol III alpha C-terminal sequence was proposed to provide the binding site for beta2 (Lopez de Saro et al. (2003) EMBO J 22:6408; Leu et al. (2003) Mol Cell 11:315). We revisited the issue using a combined genetic, function and biophysical approach, and showed that the initially identified internal site was solely required for beta2 binding, processivity and replication in vivo (101). Subsequent structural and genetic studies have confirmed our conclusions (Wing et al. (2008) J Mol Biol 382:859; 129).
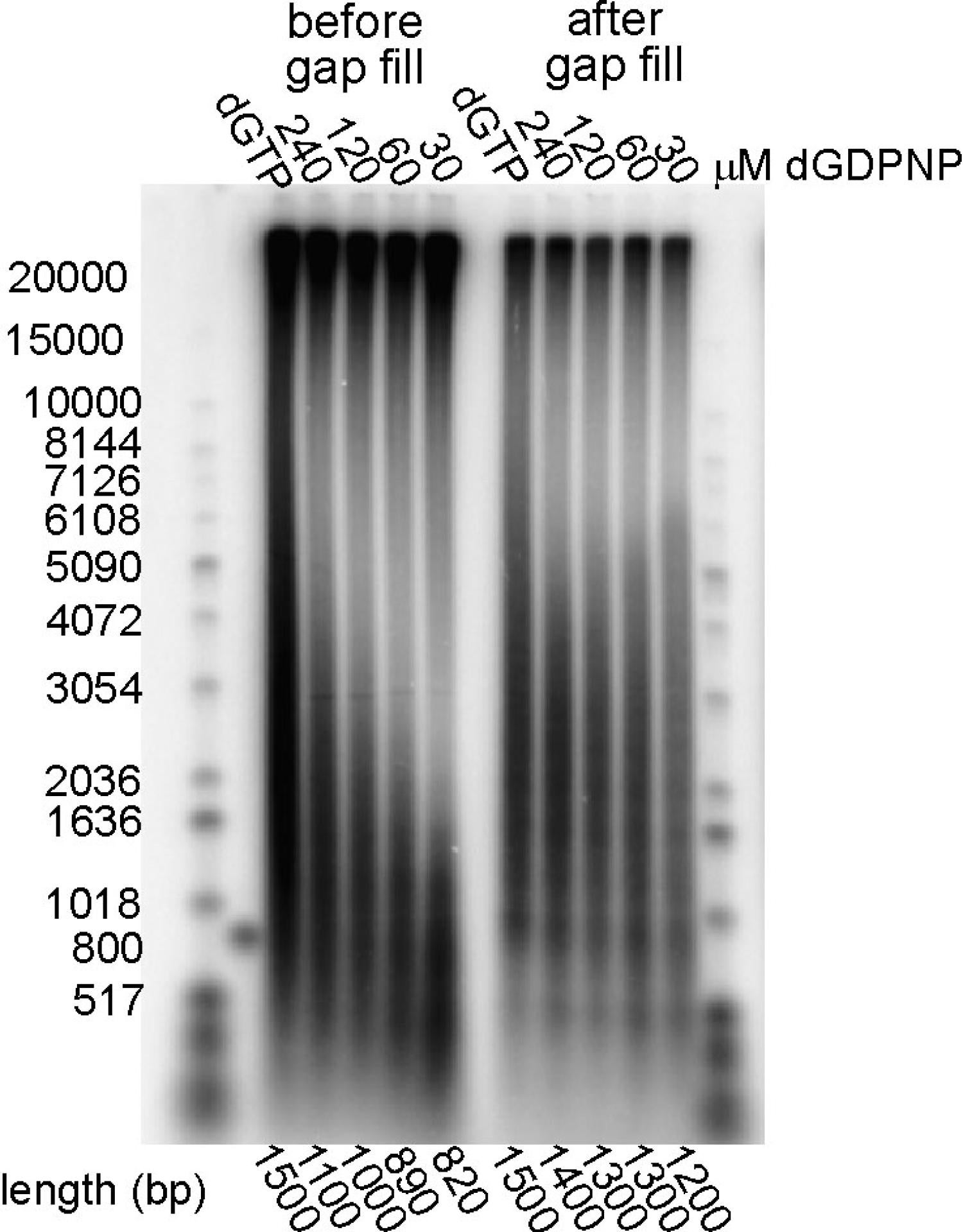
In our final activity in this area, we tested two models for Okazaki fragment cycling—the Marians signaling hypothesis and the collision hypothesis, that proposed running the lagging strand polymerase into the preceding Okazaki fragment triggered recycling. Model studies measure the kinetics of release upon elongation and collision showed that release was 1000-fold too slow to support the kinetics of lagging strand synthesis (115). We next took advantage of a synthetic mini-circle with asymmetric GC distribution and dGDPNP to selectively slow the synthesis of the lagging strand. At slower lagging strand elongation rates, we observed shortened Okazaki fragments with gaps between them, consistent with the signaling hypothesis and contrary to the collision model, where increasingly longer fragments would be expected (121). We showed it was the availability of a new primer, not the action of DnaG per se, that triggered cycling (121).
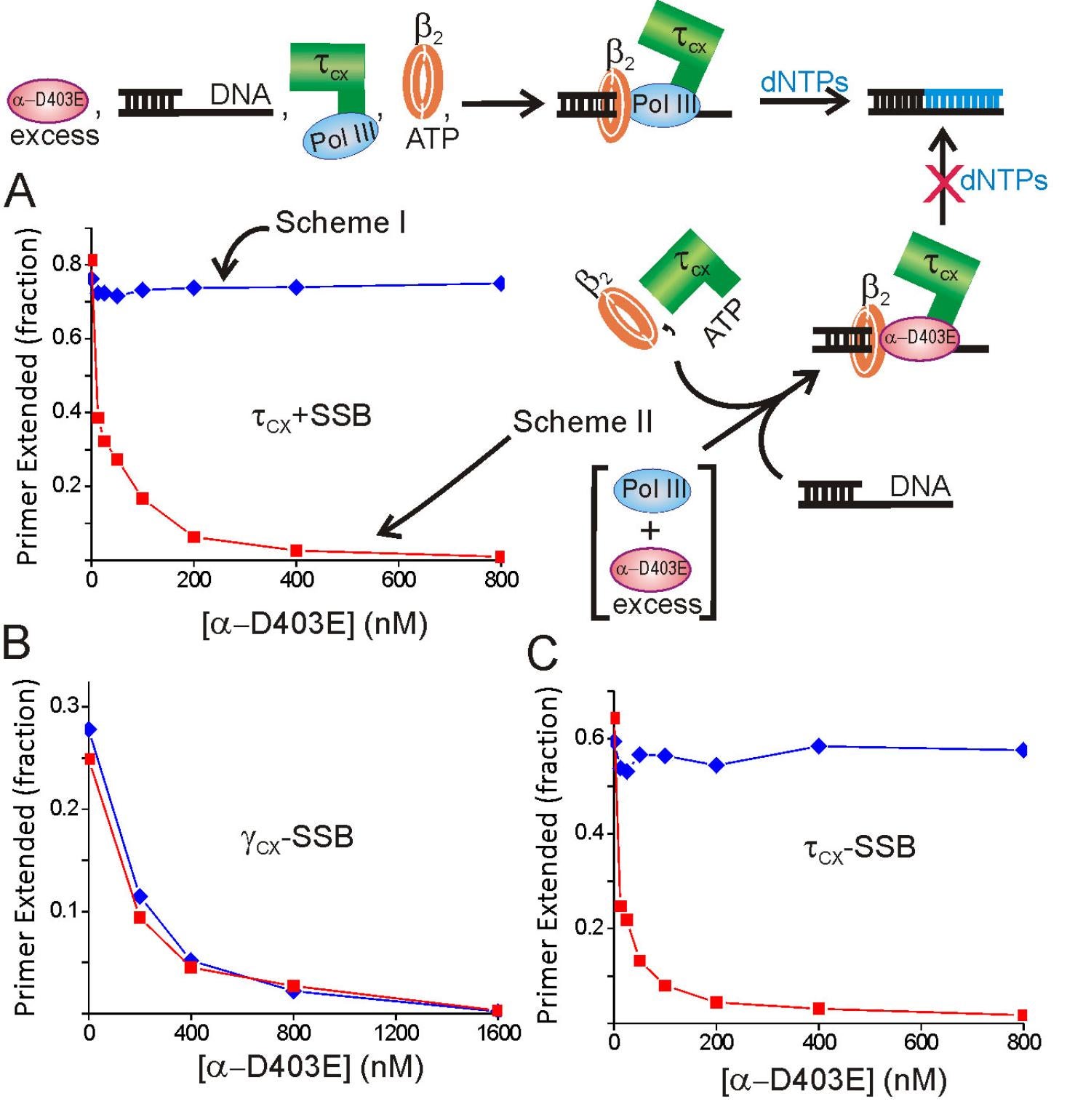
Mapping interactions within the Pol III HE at the protein domain level: We mapped most of the known Pol III HE interactions down to the domain level (71,72,77,80,81,83,86,87,88,92,93,95,97,103). Most of these interactions were within the enzyme free in solution. Knowing how these interactions change and what new interactions form at each stage of the Pol III HE reaction is essential for understanding how replicases function and represents a deficit in current knowledge. For example, an SSB-Pol III HE interaction occurs with a site distinct from the known SSB-chi interaction during initiation complex formation (113).
Highly Processive Thermophilic Replication System: We have discovered a novel highly processive thermophilic replicase that, coupled with replication fork proteins, should be capable of replicating entire chromosomes and megabase segments of DNA (76,98). We also demonstrated that the PHP domain of Tth Pol III alpha contains a novel Zn++-dependent nuclease activity (104). Preliminary evidence suggests that the E. coli PHP domain has a similar activity (111). Testing the generality of these findings and their importance to replication fidelity remains an area for further investigation.
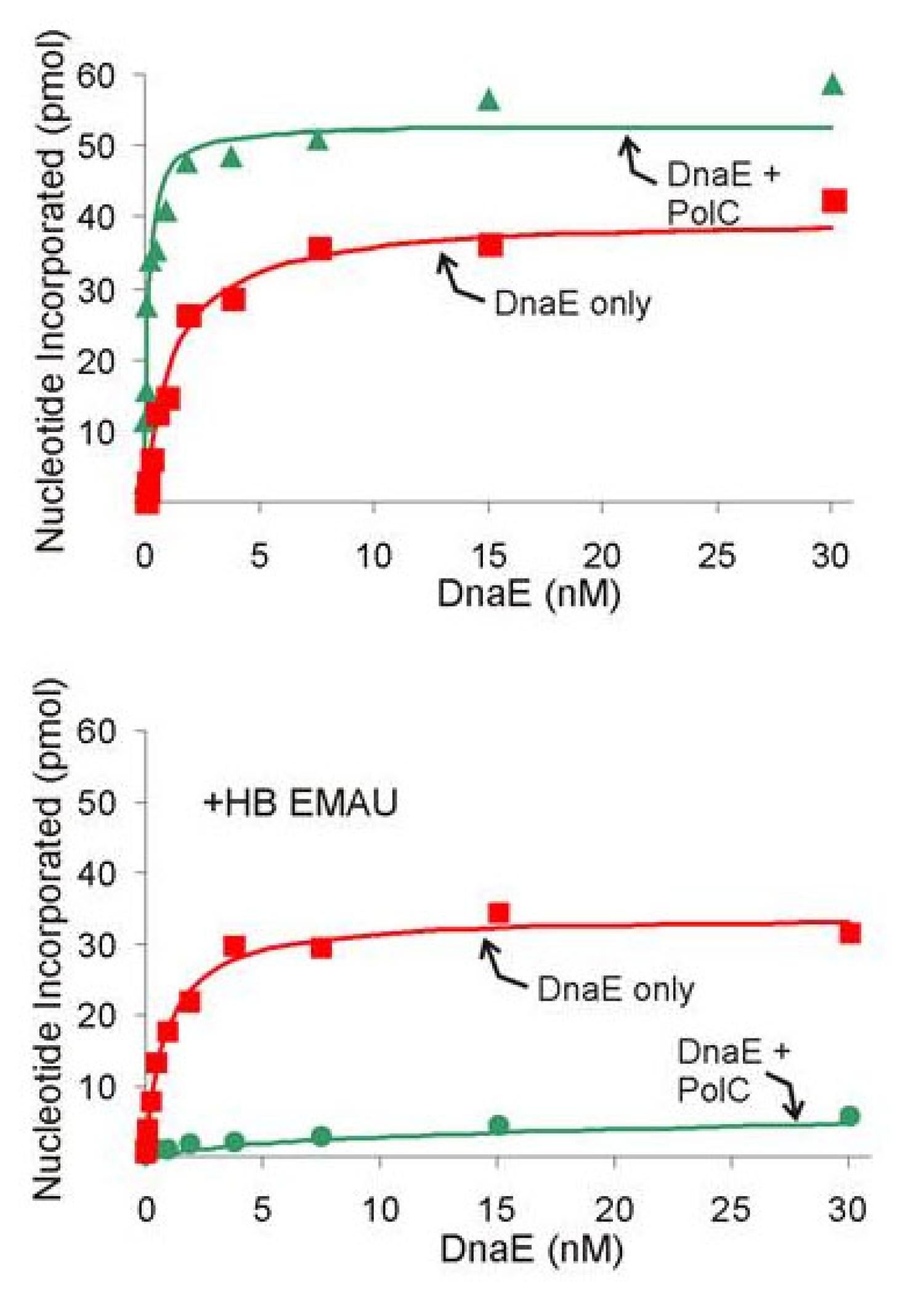
B. subtilis DNA Replication: The low GC Gram-positive bacteria diverged from E. coli approximately two billion years ago. This model organism provided an opportunity to learn how the central themes of DNA replication vary between highly divergent organisms. We established a full in vitro system that requires 13 purified proteins (107). A key difference from the long-standing E. coli model is that two DNA Polymerase IIIs participate in lagging strand replication. One, DnaE, elongates RNA primers and hands off the product to PolC for further elongation (107). DnaE is highly processive by itself, so the handoff occurs by an active displacement. This is analogous to the functioning of Pol alpha and delta in synthesis of the eukaryotic lagging strand. Using this system where handoff occurs frequently and rapidly offers a promising avenue of investigation that may provide general information pertaining to the broader problem of error-prone polymerase exchange at the replication fork. Collaboratively, with Silvia Ayora, we reconstituted a 11-protein system for subtilis-phage SPP1 that employs SPP1 origin-binding protein, helicase, helicase-loader and SSB and host primase and polymerase components (118). This system shows close similarities to the coli-phage lambda replication system established by Roger McMacken’s lab (Mensa-Wilmot et al. (1989) J Biol Chem 264:2853).
Chemical Biology of DNA Replication: Providing a bookend for a career that began with employing chemical biology (before the term was coined), one of our final efforts was directed toward developing chemical biology approaches for studying DNA replication. We developed methods for discovery and development of small molecules that block each of the steps of the complex replication process. We published a demonstration project from my academic lab (109,127), but the main effort took place within a company, Replidyne (RDYN), for which I was the scientific founder. Successes within the company included developing efficient methods for screening nearly all of the known bacterial DNA replication targets and methods to deconvolute the target of inhibitors discovered in multi-component assays, that culminated in finding compounds that served as useful antibacterials in animal models. Unfortunately, the company became re-directed toward an in-licensed compound that led to its failure. After my departure, concomitant with in-licensing, lack of cooperation and ill-directed protection of information prevented publication of many important scientific discoveries.
This brief format did not permit giving credit to the numerous students, postdocs, professional research assistants and other lab members who did most of the work I describe. Their intelligence, skill, and hard work made this work possible and their enthusiasm, discussions, good nature and development as scientists made my career gratifying and enjoyable. I extend my deepest gratitude to them.
I retired, then closed my research laboratory on April 30, 2016.