
Atomistic Hydrodynamics and the Hydrophobic Effect in Two-Dimensional Crystals
By some estimates, nearly 80% of the world lives with water insecurity. Water desalination is a method of generating potable water that is free from rainfall constraints, but with current membranes, desalination is an expensive and energy intensive process. Two-dimensional crystals, like graphene, hold unique promise for ultrahigh throughput semipermeable membranes in water desalination, but the water transport mechanisms through them remain poorly understood.
Using porous graphene as both a specific model system and an analog to other two-dimensional crystals, our group has developed theory, simulation, and analysis techniques to understand how hydrophobicity impacts aqueous transport in these materials.[1] The central problem that theory faces is one of vastly different competing scales. The gradients in pressure and density that drive flow are macroscopic, but the pores embedded in sheets that exclude ions with high fidelity are so small that they only admit about two water molecules at a time. Thus the motion of a few molecules bottlenecks the macroscopic current. This disparity in interacting scales challenges continuum hydrodynamic models, which are trustworthy away from equilibrium but are dubious at atomic length scales.
Drawing on foundational principles in equilibrium statistical
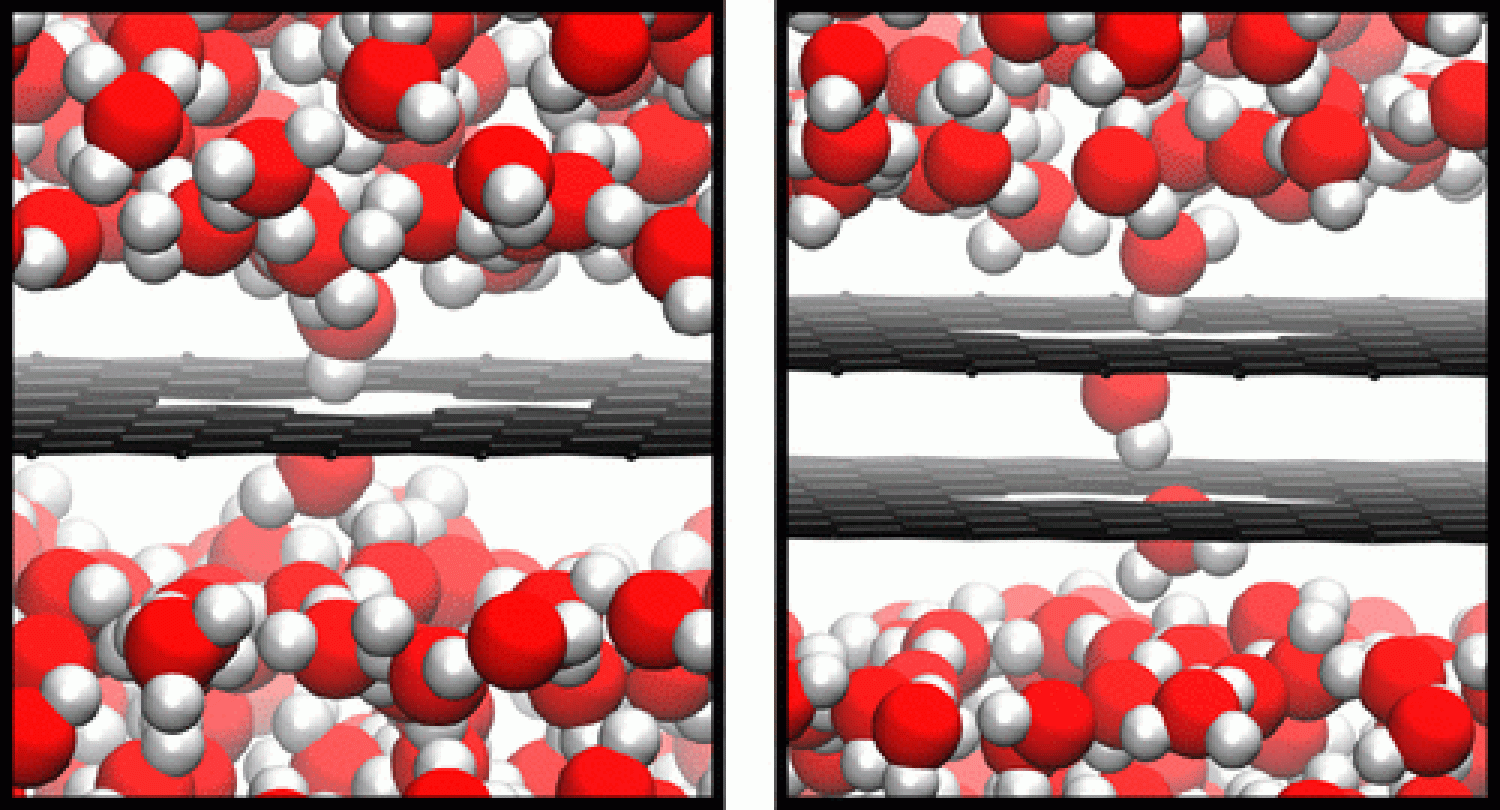
Our Gaussian Dynamics method is quite general and allows access to a host of problems where both hydrodynamic and atomistic degrees of freedom are important but have remained difficult to address at microscopic scales using computer simulation. We are currently using Gaussian Dynamics to study capacitive desalination, crystal nucleation under fluid flow, and friction-induced melting of ice.
- Strong, S.E. and Eaves, J. D., Atomistic Hydrodynamics and the Dynamical Hydrophobic Effect in Porous Graphene. Journal of Physical Chemistry Letters, 2016. 7(10): p. 1907-1912
- Ostrowski, J.H.J. and Eaves, J. D., The Tunable Hydrophobic Effect on Electrically Doped Graphene. Journal of Physical Chemistry B, 2014. 118(2): p. 530-536