
Projects
Proteins bind to the proteasome through ubiquitin receptors
The ubiquitin-proteasome system is essential to eukaryotic life through the regulation of protein degradation. Ubiquitinated proteins bind to the proteasome via ‘intrinsic ubiquitin receptors’ – subunits of the proteasome cap which promote docking of ubiquitinated substrate to the proteasome for subsequent degradation. In animal models, these subunits are essential to proper development and survival. In addition to intrinsic receptors, mammals also express ‘extrinsic ubiquitin receptors’; these proteins are not subunits of the proteasome but are capable of simultaneously and reversibly binding ubiquitinated proteins and the proteasome.
Ubiquilins facilitate proteasomal degradation of certain proteins
The largest family of extrinsic ubiquitin receptors are called Ubiquilins (Ubqlns), which facilitate degradation of a poorly defined subset of ubiquitinated proteins. The 5 Ubqlns all contain ubiquitin-like (UBL) and ubiquitin-binding (UBA) domains, but the tissue expression pattern of each Ubqln is unique. Since their discovery, multiple Ubqlns have been implicated in cancer and neurodegenerative disease. In particular, mutations in Ubqln2, which is highly expressed in neurons and muscle, were recently identified as the cause of an X-linked, dominant form of familial Amyotrophic Lateral Sclerosis (fALS). These findings have spurred renewed interest in understanding how Ubqlns promote cellular health, and the two questions that are central to the understanding Ubqln biology are the identity of Ubqln client proteins and the consequences of client protein dysregulation.
Ubiquilin2: mutations cause ALS in humans
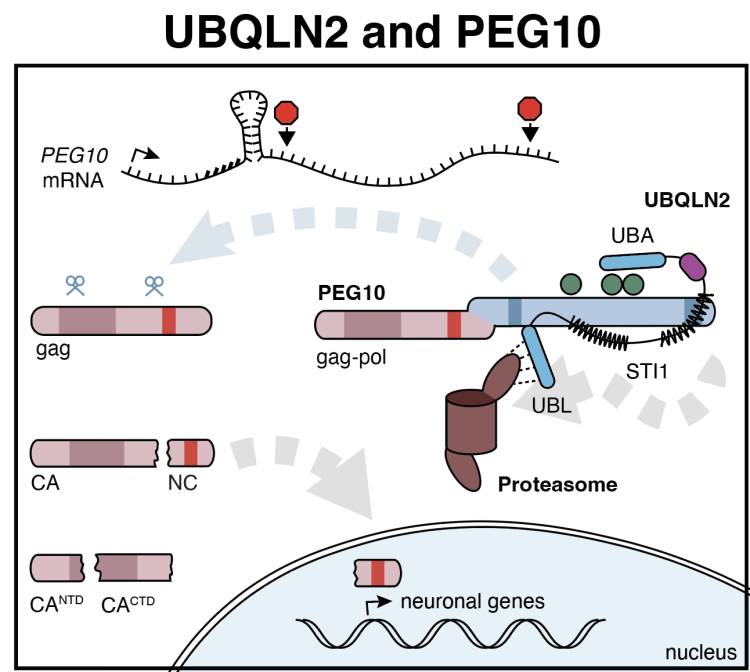
In 2011, it was found that mutations in Ubqln2, which is highly enriched in neuronal and muscle tissue, cause a familial form of Amyotrophic Lateral Sclerosis (ALS) with Fronto-Temporal Dementia (FTD). While ubiquitinated agregates of Ubqln2 protein are visible in brain tissues of these patients, it was unknown how the perturbation of Ubqln2 function leads to disease. Whole proteome analysis of animal models of Ubqln2-mediated neurodegenerative disease recently identified a small number of proteins which are significantly in CNS tissues upon Ubqln2 perturbation. Surprisingly, two of these proteins are from the same family of retroelement-derived proteins: PEG10, and RTL8.
These findings raise two central questions:
- What makes PEG10 depend on UBQLN2 for its degradation? The characteristics that define Ubqln clients are still largely unknown: some studies have classified proteins based on their hydropathy, others on their folding state. As such, there is no unifying model for what makes a protein depend on Ubqlns for their degradation. Using PEG10 as a model client, we are determining the biochemical characteristics that render it uniquely dependent on Ubqln2 for its degradation.
- How does accumulation of PEG10 lead to neurodegenerative disease? Ubqln2-deficient mice develop age-dependent neurodegenerative disease, which correlates with the dysregulation of Ubqln2-dependent client proteins. How does the dysregulation of PEG10 contribute to disease? Here, we are using a combination of cell-line and primary neuron model systems to explore how PEG10 abundance and self-cleavage influences neuron health.
Ubiquilin1: essential to proliferating cells under very specific conditions
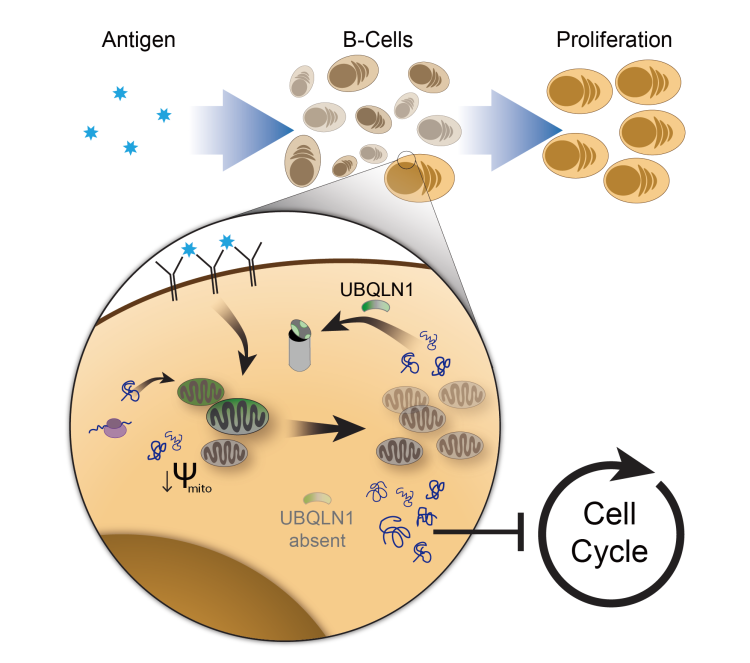
Ubqln1 is widely expressed in tissues and cell lines, but its precise role in maintaining cellular health remained elusive until recently. Unlike many cell culture systems where the loss of Ubqln1 is tolerated, we have found that Ubqln1 is uniquely necessary for B cell proliferation downstream of BCR activation. In addition, we recently discovered that Ubqln1 can be made essential to HeLa cells, where its loss is normally tolerated, upon specific cytokine treatment. In both cases, stimulation of the cell has revealed a unique susceptibility to Ubqln1 loss resulting in a block in nascent protein synthesis and subsequent cell proliferation.
These findings raise three important questions:
- What are the clients leading to these phenotypes in very different cell types? For stimulated B cells, an accumulation of mislocalized mitochondrial proteins is thought to occur downstream of Ubqln1 loss, leading to the block in protein synthesis. However, recent work shows no change to mitochondrial proteins in our Ubqln1-sensitive HeLa cell model. What are the true clients of Ubqln1? And why do they differ in each model system?
- How does the accumulation of Ubqln1 client proteins lead to a block in new protein synthesis? Accumulation of proteins in the absence of Ubqln1 expression is followed by a block in nascent protein synthesis; however, the mechanism of this block is unclear and appears to be independent of well-defined regulatory pathways such as the UPR. Instead, both model systems show an upregulation of innate immune responses linked to protein synthesis. Current projects in the lab are focused on identifying the mechanism of this translational block.
- How do these findings inform our understanding of malignancy and immunotherapy? Given that Ubqln1 depletion limits B cell and HeLa cell proliferation, perturbation of Ubqln function could have similar effects on malignancies. In addition, the upregulation of innate immune responses upon Ubqln1 loss may improve the success of cancer immunotherapies, which depend on immune activation for cancer targeting. The laboratory is using both in vivo and in vitro approaches to investigate these questions.